

欧洲药品管理局(EMA)是生物类似药监管的先行者,它的一举一动对全球范围内的监管行为都有示范作用。早在2001年的指令2001/83/EC中就有相关生物类似药的申报要求,随后的2005年,EMA正式颁布一系列指南供业界参考,并于2006年批准了第一个生物类似药-来自于Sandoz的重组人生长激素。了解欧盟的监管情况,不但是产品上市欧盟这个大市场的前提,对于预见美国或其他国家的监管行为也是十分有帮助的。
一、法规情况
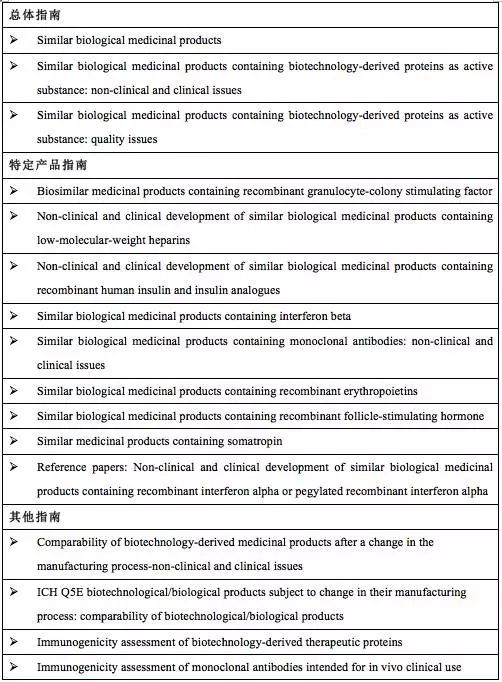
在EMA的官方网站上,罗列了所有有关Biosimilar的法规指南。这些指南共分为三类:总体指南(包括生物类似药的概念,质量、临床前和临床方面的总体议题),针对特定产品的临床前和临床指南(如胰岛素、促红细胞生成素、单克隆等产品或类型)和其他相关指南(包括工艺变更和免疫原性评价方面的内容)。可以看出,这些指南既全面又有针对性。并且,随着实践的深入,这些指南从发布之日起,都会不定期修订。
二、申报文件、申报路径和审评过程
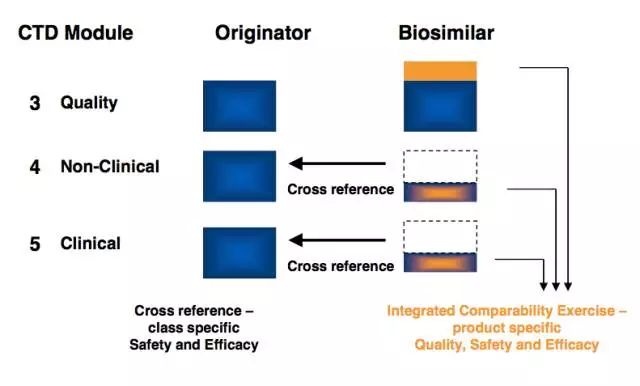
申报文件的格式要求是CTD。质量方面,除了传统的产品本身的CMC数据外,还需要全面的对比数据。非临床和临床方面,需要由根据相关指南设计试验而得出的数据。
申报路径上,对于Biosimilar产品只允许申请集中审评程序(Centralized Procedure, CP)。CP的审评周期是210天,但这不意味着210天就可以得出审评结论。因为如果在第120天,EMA发出缺陷信,审评时钟就会暂停,待其收到缺陷信回复后,重新开始计时(即开始第121天的计时)。
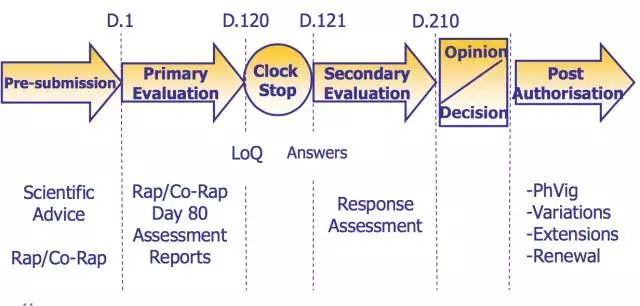
值得一提的是,是否批准Biosimilar产品由EMA决定,但已批准的产品是否可以替换原研产品由各个成员国国家自行决定。
三、批准情况
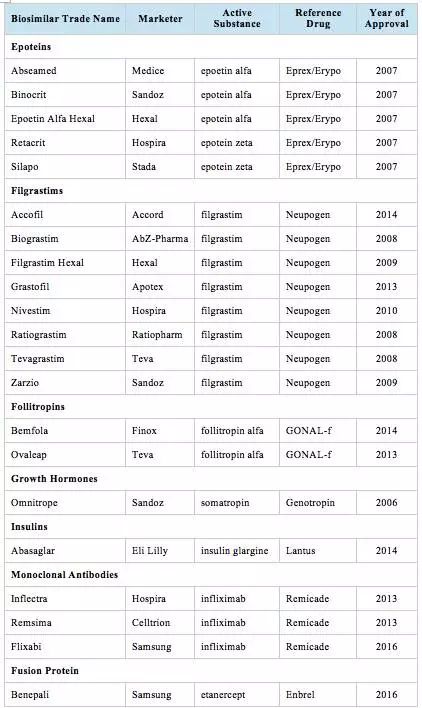
相比美国FDA,EMA的成果是丰富的,并且富有勇气和开拓精神。截至目前为止,共有7类21个品种获得了批准,具体情况如下:
四、总结展望
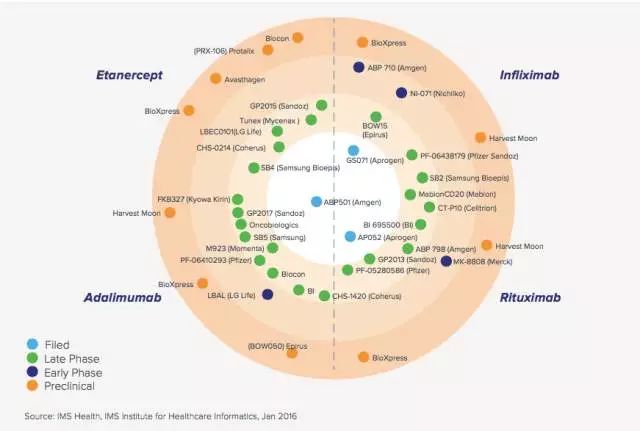
下图是截至今年1月份,全球Biosimilar产品的研发概况。业界开发Biosimilar产品的热情不减,药政当局的实践就会不断深入,业界和药政当局的博弈也会精彩纷呈,让我们拭目以待吧。
参考资料
EMA网站:Biosimilarsin the European Union-regulatory perspectives, Kuala Lumpur


