

摘要
QbD的起源
QbD是什么
QbD应用于工艺开发
i. 确认CQA
ii. 定义产品设计空间
iii. 定义工艺设计空间
iv. 定义控制策略
v. 描述产品特征
实施QbD的动力
通过QbD进行一个仿制药产品的下游工艺开发的案例研究
i. 确认CQA
ii. 定义产品设计空间
iii. 定义工艺设计空间
iv. 定义控制策略
v. 描述产品特征
实施QbD的益处
结束语
QbD目前越来越受到制药行业的关注,大多数主要生物制药企业在实施QbD的不同阶段。在此本文列举了一个通过QbD的方法逐步进行工艺开发的例子——GCSF仿制药纯化工艺开发。本文将重点讨论基于工艺开发时QbD与传统方法相比的优势,并通过个案分析帮助那些在想在生物制品工艺开发和商业化生产时实施QbD的人们。
QbD在生物制药行业的作用日趋突出,该行业要遵循美国FDA颁发的创新医药制造业PATA框架和质量保证文件。其所包含的ICH指导原则中的相关概念得到了全球的普遍认可,ICH Q8 药物开发,ICH Q9质量风险管理,ICH Q10药物质量体系,ICH Q11原料药开发和生产。在过去的十年里,生物技术行业和其监管机构尤其是美国FDA实施了必要的基础工作来为QbD的实施铺平道路,并解决了多种包括可能影响其成功实施的问题。虽然到目前为止仅有一个生物药用QbD的方式通过了申请,但其相关的工具和方法已经成为全球化制药公司的工作过程中的固有形式。
本文将列举生物仿制药GCSF进行逐步纯化工艺开发的例子对QbD进行诠释,并通过案例分析帮助那些在想在生物制品工艺开发和商业化生产时实施QbD的人们。
在ICH Q8中对QbD进行了定义,即在可靠的科学和质量风险管理的基础上,预先定义好目标并强调对产品与工艺的理解以及工艺控制的一个系统的研发方法。传统方法是厂家确定目标产品定义工艺,然后针对产品的期望结果进行检测。如果产品质量达到预期,厂家就会固定工艺,然后在很小的工艺操作范围内进行生产。这就是所谓的“如果操作的过程是一致的,那么生产的产品也应该是一致的”,这种方法已经被生物制药公司应用了30多年了。而QbD方法是针对生物药的开发与生产,与上面描述的传统方法有着显著的差异,结果说明见图1。关键步骤:(1)确认对安全性和有效性有显著影响的产品属性(例如QTPP和CQA);(2)通过工艺设计来实现这些属性;(3)通过稳健的控制策略来保证工艺性能的稳定;(4)对工艺进行验证和文件化来证明控制策略的有效性;(5)继续在产品的生命周期内进行监测,来保证稳健的工艺性能。此外,通过知识管理、风险评估和管理、原料管理、统计学方法以及过程分析技术来为QbD的实施提供基础。
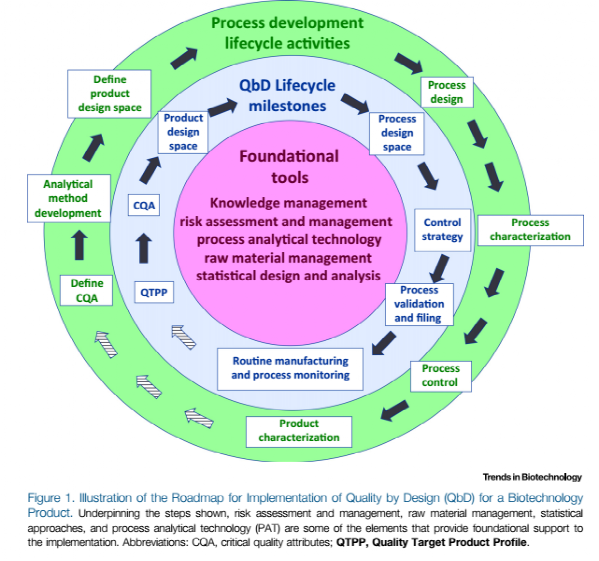
图1说明了QbD也可以叫做工艺开发,具体将在下面进行进一步的描述。
将CQA定义为“物理、化学、生物或微生物学属性,或是对特征进行适当的限定、范围或分布进行描述来保证目标产品质量”。由于工艺开发应当关注于实现目标产品CQA定义的质量达到想要的一致性,因此在工艺开发前就需要确认产品的CQA。在产品开发的所有关键阶段都需要对CQA名单进行回顾,以便保证非临床、稳定性和临床研究的数据更加可靠。当这些发生时,需要进行工艺能力评估。
“设计空间”这个概念作为一个工具已经普遍的应用于生物药QbD的实施。ICH Q8将其定义为:输入变量(比如物料属性)的多维组合和交互作用以及已被证明的能够为产品质量提供保证的工艺参数。只要是在设计空间内进行的操作,不认为是变更。而在设计空间外的操作才被认为是变更,并且一般情况下要通过监管机构批准工艺变更后才能使用。设计空间是由申请人提议的,并且要受监管的评估与批准。
一旦确认CQA,下一步就要定义CQA变量的可接受水平。设计空间窗口的大小直接影响工艺开发,如果产品设计空间较小(标准要求比较严),那么工艺参数同样要控制在较小的操作范围内来保证产品能够达到标准。相反,如果产品设计空间较大(标准要求宽松),那么工艺就相对灵活一些。
在定义CQA和可接受的变量范围后,应进行工艺特征研究将CQA和工艺每一步的工艺变量、原料属性和进料属性相联系。在QbD范例中通过DoE方法来进行研究的,这样不仅可以明白变量的主要影响,而且也可以弄明白它们间的交互作用。研究结果在复合、多维和经验的模型形式中是比较典型的,而该模型能够将不同的CQA和多种因子相联系。
控制策略定义为:源于对现行产品和工艺的理解,能够保证工艺性能和产品质量的有计划的一些列控制活动。在QbD中,控制策略要围绕着规定工艺能力和产品CQA。上述的经验模型能典型的用于定义有效的工艺参数的可接受范围。然后通过控制策略来保证工艺能够按既定轨道进行,并且产品的质量能够始终如一的满足预定义的质量要求。
一旦定义并实施了控制策略,就要进行适当的产品质量监控来证明设计的工艺具有稳定的生产出能达标产品的能力。这一活动也是对QbD能用于工艺开发的验证。
实施QbD具有如下几个动力:
(1)QbD中的操作范围是比较宽泛的,这来源于经确认的影响CQA的关键工艺参数的鉴别。这与传统的制造业形成鲜明对比,由于工艺开发者并不知道哪个工艺参数是关键的,传统制造业的所有工序的操作范围是比较窄的。(2)在QbD中产品质量参数范围是比较宽泛的,这是因为产品的质量是基于对工艺和产品的理解而制定的。这促使我们的操作具有一定的灵活性,特别是对非CQA而言,而这种灵活性主要体现在比较宽的操作范围。
(3)更稳健的工艺。以动态控制策略为基础的过程分析技术能开发出更稳健的工艺,该工艺能够实现目标产品的质量是始终如一的。
(4)显著降低失败风险,因为在QbD中我们是按照PAT范例进行实时或接近实时的产品质量测试的。
(5)工艺变更负担小。QbD的监管机构备案文件是知识的基础,它关注于阐述工艺和CQA以及CQA和产品的安全性和有效性的关系。而传统的备案文件要罗列出所有参数和属性,包括CPP和非CPP,CQA和非CQA。因此,当需要做工艺变更时,我们的备案文件也需要做变更。这给生物技术企业带来巨大的负担,这也是为什么企业都倾向于保守并且拒绝接受新的技术和实践。
(6)更少的生产偏差。有效的生产能将QbD贯穿于产品的整个生命周期中,这使得操作范围和质量参数比较宽泛,从而减少生产偏差。由于生产商已经有数据对偏差对产品质量的影响进行了阐述,因此当出现任何与前面所描述的数据不一致时我们可以相对迅速的将其解决。
(7)得到更理想的工艺。在试验过程中广泛的使用DoE进行优化,并考虑生产力和经济学,这能够开发出比较理想的工艺。通过DoE选择的条件是全局最优条件,而不仅仅是局部的最优条件。因此工艺是比较稳健的,并且可以避免不必要的昂贵的返工。
下面本文将结合下面给出的案例浅谈一下怎样才能实现以上描述的益处。
通过QbD进行一个仿制药产品的下游工艺开发的案例研究

在本章节中将会提供一个案例来说明QbD是如何在生物仿制药(GCSF)下游工艺开发中实施的。本章节的案例来自于对先前已经发表过的研究的总结。
为了确定GCSF的CQA,我们进行了全面的文献检索、GCSF商业生产商的调查以及现存的GCSF的相关著作,结果如表1。表1中,CQA能够分为几类:产品相关变体杂质、宿主细胞杂质、设施相关属性和与生物治疗药建立同一性和效力相关的东西。
产品相关的变体和杂质是一类重要的值得进一步讨论的分类,对于GCSF而言需要进一步考虑的关键属性包括产品的氧化、还原、聚体和fMet的形式。有人认为蛋氨酸的氧化能导致蛋白的物化性质改变,从而使蛋白失去生物活性。同样的,GCSF的还原形式同样被认为是对其生物活性有影响的。此外异源蛋白的高水平表达已经被证明了其能够导致GCSF N甲酰蛋氨酸变体形式的甲酰基团部分保留。但是与天然GCSF蛋白相比,我们认为变体是有相同的生物活性的。它的存在可能会引起病人的免疫原性反应,因此需要监测和控制其含量。最后,聚体被普遍的认为能够引起病人的免疫原性反应,因此将其作为生物治疗药的CQA考量。正如表1描述的那样,首先应该关注的是能够被下游工艺影响的CQA。
本案例中关注的是生物仿制药产品的下游工艺开发,首先通过文献检索来建立CQA。如果是进行开发创新要的话,过程将会有所不同。在本案例的基础上,应当通过其它方法进行重要的补充。经常的,该策略应当涉及以下几方面的风险评估:分子(热点和分子工程)设计;产品和其它平台产品通过体内和体外实验进行非临床研究;产品和其它平台产品的临床研究;发表的文献。风险评估的结果应该是危急程度的连续统一体,产品质量属性应该罗列其中。对安全性和有效性有影响的(比如聚体种类)应当排在高危急程度位置,对安全性和有效性没有影响的(例如C末端赖氨酸杂质)排在低危急程度位置。
表1罗列了本案例选择的产品质量参数,这主要是基于GCSF专著提供的相关信息。这些数据将会进一步用于下游工艺的设计。
本步骤是本案例中关注的重点,涉及了多个步骤方法的应用,包括:复性工艺的优化;建立高通量工艺开发平台应用于层析工艺开发;多种层析模式的评估;工艺整合。本步骤的关键点是为每个工序建立一个合适的缩小的规模,该规模在使用前必须保证是有效的,以便保证在研究过程中产生的数据能适用于商业化规模生产。
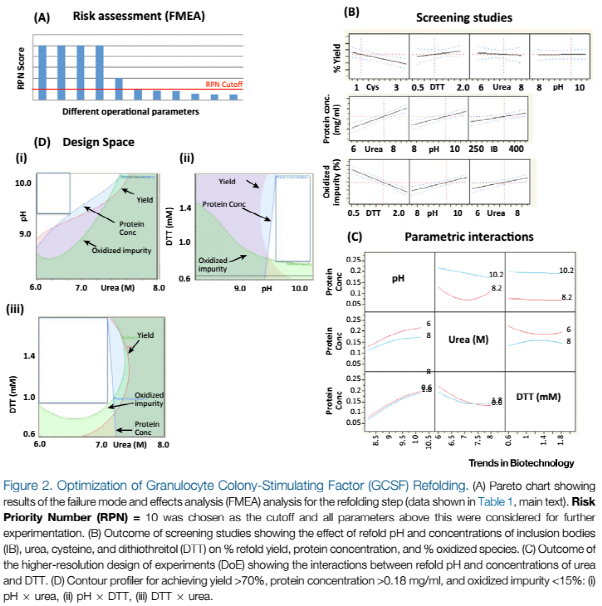
我们将使用基于QbD的DoE方法进行复性工艺优化。首先我们通过风险分析来确定需要进行试验考察的工艺参数(图2A)。接下来,我们通过部分因子初筛试验设计(图2B)来考量温度;复性pH;包涵体、尿素、半胱氨酸以及DTT的浓度。基于以上研究结果,我们将使用全因子DoE试验对pH、尿素浓度以及DTT浓度做进一步考量,来阐述其对每一步得率和产品质量的影响,以及他们之间的交互作用(图2C)。结果本工艺的复性率能达到77 %,这远高于商业化的GCSF的复性率(40-60 %)。最终将产品的设计空间设定为可接受的范围:得率大于70 %;蛋白浓度大于0.18 mg/ml;氧化的杂质小于15 %;尿素浓度6-6.6 M;pH 9.5-10.2;DTT 0.92-1.8 mM。设计空间的说明见图2D。高回收率和CPP宽泛的设计空间能够体现遵循QbD的方法进行工艺开发的价值。
接下来的工作是建立复性后的纯化工艺。通过商业调查我们发现,目前应用于商业化生产GCSF的工艺模式主要是:阳离子交换层析、阴离子交换层析、疏水复合模式层析(按受欢迎程度下降的顺序)。每个层析步骤能够使用结合-洗脱模式或流穿模式。基于之前的知识和商业调查,我们决定关注用于GCSF纯化的阳离子交换层析、阴离子交换层析以及复合模式层析进行评估,结果见图3。
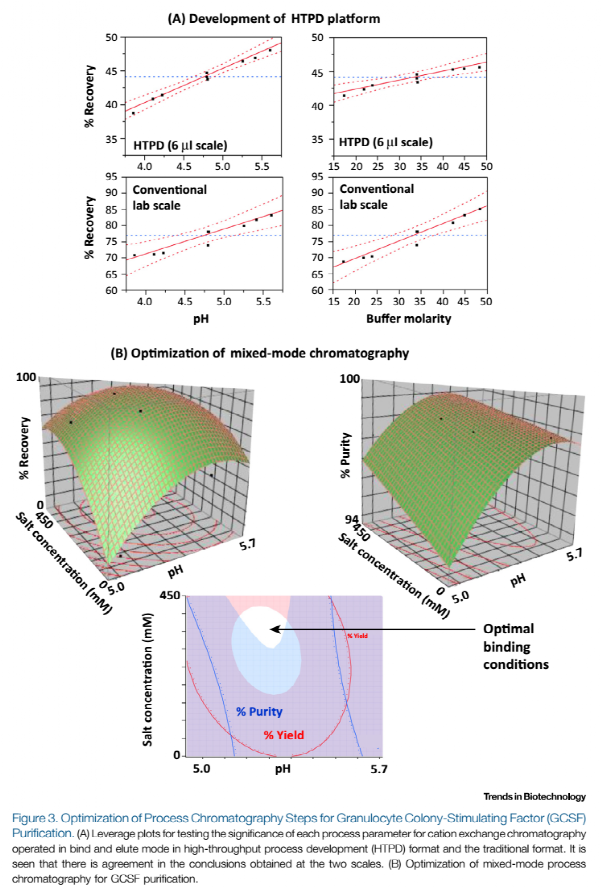
考虑到需要做大量的层析试验,我们开发了高通量工艺开发平台。为了达到这个目的,我们选择了GE医疗的Predictor PlateTM进行试验。我们发现手动操作2-6 μl规模存在一定的挑战,因此我们需要开发一个能够有效降低这种挑战的方案,最终我们将HTPD平台和传统的实验室0.5 ml规模的结果进行对比,结果表明HTPD平台能够成功的用于工艺开发。虽然HTPD平台中每一步的收率的绝对值较低,但是其趋势是正确的。因此该平台能够应用于半定量评估多种输入参数的影响(图3A)。在实际应用之前我们认为该模型是具有统计学意义的。但通过进一步调查研究发现,造成每一步收率低的原因是HTPD膜板对GCSF有非特异性吸附。而且其它一些疏水作用不强(比如单克隆抗体)的蛋白的非特异性吸附作用是可以忽略的。
一旦HTPD平台开发成功,我们能将其用于多种层析填料和不同模式下的操作条件进行快速筛选。首先,我们通过HTPD平台对5家主要供应商的9种不同的阳离子交换层析填料进行筛选。通过DoE的方法对每种填料的最优条件进行优化,包括pH和缓冲液摩尔浓度。之后要在传统实验室规模进行洗脱策略的优化。结合-洗脱模式的阳离子交换层析GCSF纯化的最优结果表明,其最高回收率能达到80 %。相似的,我们通过HTPD平台对4家主要供应商的8种不同的阴离子交换层析填料进行结合-洗脱和穿透模式筛选。结果表明,在结合-洗脱模式时,阴离子层析的收率小于30 %。这主要是因为在低pH(译者认为应该是高pH,结合是在高pH,穿透模式是低pH)下GCSF非常不稳定。而在穿透模式下,产品收率几乎达到100 %,并且宿主细胞杂质显著降低(DNA约0.5个log降,HCP 大于1个log降)。最后,通过HTPD平台对复合模式层析填料进行评估。HTPD平台结合传统实验室步骤对每种填料进行优化,最终仅使用复合形式填料就能使收率达到90.0 %,同时纯度大于99.0 %(图3B)。这很可能是因为通过pH和盐浓度进行梯度洗脱的疏水离子复合层析对相关产品的变体的分离有特异选择性。
因此,最终优化后的工艺为:通过pH调节进行复性——一步疏水离子复合层析。
控制策略是QbD的关键组成部分,同时也是过程分析技术的基础。在本案例中,控制策略涉及受下游纯化工艺影响的产品相关的CQA,即氧化、还原和聚体种类。在复性和纯化工序中,通过监测产品质量来评估分析工具的实用性。包括:RP-HPLC、傅里叶变换红外光谱、紫外圆二色谱、SEC-HPLC、荧光。考虑到灵敏度、精度、分析时间和可操作性,我们决定将RP-HPLC和SEC-HPLC用于监测GCSF复性工序,将RP-HPLC用于在线监测复合模式纯化工序的洗脱液(在线分析技术)。不同文献资料表明,产品目标CQA的一致性的显著提高是我们在实施PAT策略时所愿看到的。
QbD范例中生物药工艺开发是建立在产品质量始终如一的基础上的。产品质量是主要的优化目标,比如:工艺的收率、生产力和生产周期。在本仿制药的案例中,产品质量的评估要与原研药进行可比性研究。在这个过程中,我们需要进行大量的可比性研究分析,将我们的GCSF与原研药和上市的GCSF仿制药进行对比。一些关键的结果见图4。
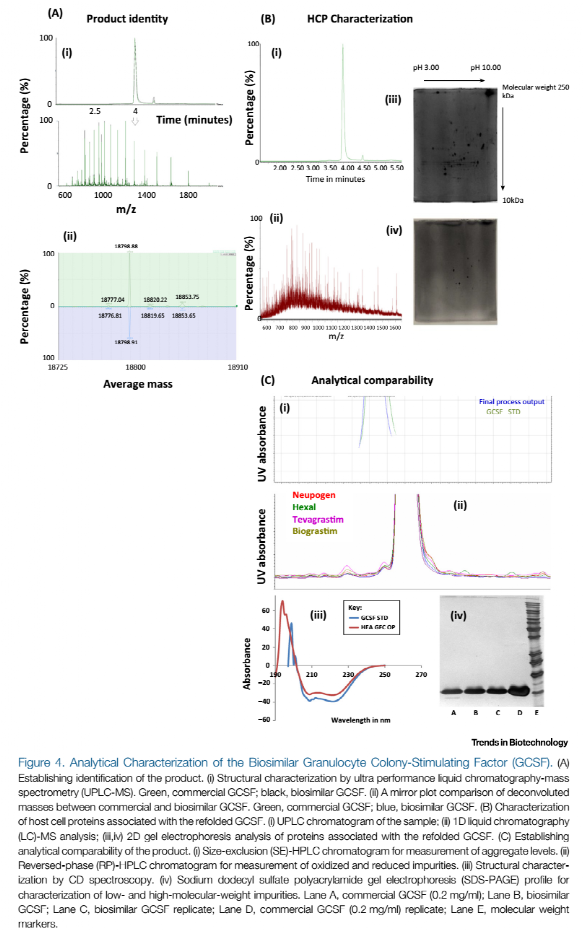
该过程的第一步是确认产品的身份,可通过质谱进行确认。图4Ai是通过复合模型柱纯化的GCSF的质谱图,图4Aii是质谱图的去卷积质谱图。结果表明,GCSF仿制药中的主要成分是GCSF蛋白,该蛋白序列中N末端带有蛋氨酸残基,但没有发生糖基化。还原和非还原消化LC/MSE肽图确定了含有N末端蛋氨酸GCSF的氨基酸序列和二硫键。这些结果表明我们的产品是GCSF,并且在整个工艺中没有发现结构变体。
下一步就是鉴定HCP(图4B),因此我们选择了二维交联凝胶电泳和UPLC-MS。我们在复性工艺产物中发现了多种HCP,我们将通过二维凝胶电泳做进一步描述。其中一些HCP能够被确定为典型的大肠杆菌表达蛋白。
最后,通过宿主分析工具进行分析可比性评估。图4Ci的SEC-HPLC表明GCSF产品和原研药产品中几乎不含有聚体(0.2 %和0.15 %)。同样的,RP-HPLC分析结果表明通过QbD方法生产的GCSF蛋白的质量与上市的原研药或其它仿制药相比不仅相似,而且要更好(图4Cii)。鉴于GCSF含有α螺旋结构,因此首要考虑的比较性质就包括在圆二色谱中208 nm和220 nm处有下降。图4Ciii可以看出两个蛋白的图谱匹配较好,这表明GCSF和市售药都含有α螺旋结构,而且没有其它结构变体。通过SDS-PAGE非还原电泳进行高、低分子量水平的相似性比较,结果与SEC-HPLC的结果相一致,仿制药和市售药的质量一致,并且几乎没有高分子量蛋白。最后,通过体外生物检测法来确定GCSF的生物活性。我们的GCSF的生物效价是市售GCSF的89.3 %,在产品规格范围之内(80-120 %)。这表明了我们的GCSF保持了结构完整性和功能完整性。
表1中的数据是通过QbD方法生产的GCSF蛋白的分析数据,表明其质量达到了市售产品的质量标准。此外通过图4可以看出生产的GCSF要优于市售产品。另外,表2比较了通过QbD方法生产的GCSF和市售GCSF的纯化工艺。通过QbD方法生产的GCSF的工艺包括:通过pH调节进行复性——过滤——复合模式层析纯化。市售蛋白工艺包括:通过换液方式(超滤/透析或凝胶过滤方式)进行复性——3到4步色谱纯化(中间还要通过超滤/透析或凝胶法换液)。QbD方法得到的蛋白纯化工艺不仅工艺收率提高3倍,而且工艺时间缩短3倍,因此生产工艺的生产力提高了近10倍。鉴于目前成本压力以及生物技术行业都在关注降低生物药生产成本,这是非常有意义的。
通过执行上述提议的工艺将会实现多个主要益处,其中一些已经在本案例中体现了。由于我们的试验数据是通过DoE试验(图2D和3B)得到的,我们能够定义更宽泛的合适的可接受范围和操作空间。虽然宽泛的产品质量规格不在本文范围之内,但是我们还是努力关注产品开发能够达到其质量规格。如上所述,我们提议了基于控制策略的动态PAT技术(通过RP-HPLC和SEC-HPLC监测GCSF的复性工序和RP-HPLC监测其复合模式纯化色谱工序),这已经展现了其显著改善产品一致性的能力,这能增加工艺的稳健性。作为上述控制策略益处的一个补充:在生产过程中产品质量就被检测了,并且最终制剂的检测项也会显著降低。原液和制剂的哪些属性需要检测最终还是要生产商去决定。如图2和图3所示,监管备案文件应包括工艺和产品之间的联系。这样监管机构就能知道哪些参数的重要的,哪些是可接受的范围。例如:蛋白复性案例中,pH、尿素和DTT浓度是关键工艺参数,基于对它们的控制能力我们规定了三个参数的合适的设计空间(图2D)。并且我们可以给非关键参数规定更宽的设计空间(比如包涵体浓度),这样我们的生产就会更灵活,并且监管的压力更小。我们希望高效的生产能贯穿于整个产品的生命周期,因为本案例中的数据明确的规定了设计空间,并且能够快速的解决生产过程中的任何不合格问题。最后,关于最优工艺的生产力和生产经济性在上文中已经进行了详细的说明,包括:生产工艺时间算短3倍;生产力提高了10倍。
总之,我希望上面的讨论能够说明通过QbD的实施是能得到很大益处的。

QbD是在2004年通过PAT指南发布时引入到生物技术行业的。在过去的十年里,在生物技术企业和其监管机构的共同努力下,对QbD实施的路线图进行了说明。当大家普遍的接受了这个概念,并且多数大的生产企业已经处在将QbD整合到各种工作中的高级阶段时,中小生产企业还在为QbD的成功实施进行不懈努力。对本案例的分析主要是为了通过展示QbD方法的益处来推广QbD。QbD的实施通常是资源集中的。本研究是两个全职等效资源的成果,这比生物技术企业在相似的工艺开发中所花费的资源要小的多。
我希望有更多的这样的研究能够分享到公共领域,因为这不仅能回答实施QbD所面临的各种问题,而且为实施QbD阐明了必要的商业驱动。


