

本文为本系列«如何决定单克隆抗体药物在首次人体临床研究时的起始剂量»的第一篇,将简明扼要地介绍如何使用NOAEL方法以确定单克隆抗体药物在首次人体临床研究时的起始剂量。本文主要参考FDA的相关指南。希望起到抛砖引玉的目的,促进在设计单克隆抗体药物在首次人体临床研究时,在保证受试者安全的前提下,给予更多的思考与创新,以降低新药研发的成本并最终惠及千千万万的患者。后续文章将简述其它方法,如MABEL方法等。
背景介绍
在开发的新型生物制药产品首次进入临床试验之前,确定人体安全起始剂量是最重要的步骤之一。在理想的情况下,起始剂量应该低,不会对人体造成任何伤害; 但也不能过低,从而减少首次接受人体(FIH)临床试验中接触无效剂量的患者人数。美国食品和药物管理局(FDA)和欧洲药品管理局(EMA)等管理机构早已发布了相关文件,以指导在FIH研究中如何选择最大推荐起始剂量(MRSD)或预期最小生物效应水平(MABEL)剂量。 FDA的许多指导原则已用于许多FIH研究,特别是小分子量的新化学实体,这个原则当然也适用于生物药。FDA指南的重点放在了临床前毒理学研究中用药剂量的未观察到不良反应的水平(NOAEL)上。然后通过应用适当的比例因子,将NOAEL转换为人体等效剂量(HED)。相反,EMA指南则强调最小预期生物效应水平(MABEL)方法,该方法考虑所有体外和体内药理毒理数据。通过NOAEL方法推导确定HED后,需要更进一步使用适当的安全系数降低剂量从而得出最大推荐起始剂量(MRSD),以保证起始剂量不会对人体造成毒性。
自上世纪80年代以来,单克隆抗体(mAb)被积极地纳入临床医学,特别是在肿瘤学和免疫学领域的应用日益广泛。然而,基于蛋白质的药物如单克隆抗体,相比基于化学药物,其安全性更不确定。例如,在一个FIH研究中, 所有使用TGN1412(一种针对CD28的超级激动单克隆抗体)的受试者中, 尽管使用了保守性低的起始剂量(从NOAEL得出并使用了较大的安全系数, 160), 都发生了严重的威胁生命的细胞因子风暴。这一不幸事件凸显了进行mAbs的FIH研究时选择最安全的,最大的起始剂量的重要性和困难。在TGN1412事件后,业界提出了在FIH研究时多种确定生物药人体起始剂量的方法。这些方法着重强调,在进行新型生物药的FIH研究时, 应该从多方面, 多角度地考察不同的药理,毒理终点,种属缩放方法和安全系数,以确定适当的人体起始剂量。
确定人体起始剂量方法
目前有多种方式及其组合来确定人体起剂量,可以大致分为如下几类。它们分别基于:
1)未观察到不良反应的水平(NOAEL)方法。
2)预期最小生物效应水平(MABEL)。基于MABEL的方法需要整合相关药理/毒理学等所有已获得数据的基础上并通过PK-PD建模(理想情况)来预测人体起始剂量。
3)最小有效剂量(MED),从产生最小药理学效应的临床前模型中确定。
4)药理活性剂量(PAD),由产生任何药理学效应的临床前模型中确定的人体起始剂量。
5)药理模型(即PK,PD或PK-PD),其中人体起始剂量是使用基于模型的方法确定的。
动物药代动力学(PK)模型, PK数据是确定人体起始剂量的基础,或者如果PK模型用于估计最终确定人体起始剂量的人体PK参数。
动物药效学(PD)模型,如使用受体占据或其他生物标志物的信息来确定人体起始剂量。
动物药代-药效动力学(PK-PD)模型, 即结合i) 和ii) 的模型
注意: 如上分类并非互不相容的, 如基于药理模型常常用于确定MABEL。在这种情况下,该方法仍被认为是基于MABEL。
最近对1990-2013年期间已发表的相关文献的调研总结了这几种方法在单抗FIH研究中使用的频率如图1。
图1. 单克隆抗体首次人体内研究中确定人体起始剂量方法的总体比例。注意:基于模型的方法包括基于PK模型,PD模型或PK-PD模型的方法。缩写:NOAEL,未观察到不良反应水平; MABEL,最低预期生物学效应水平; MED,最小有效剂量; PAD,药理活性剂量; PD,药效动力学; PK,药代动力学。
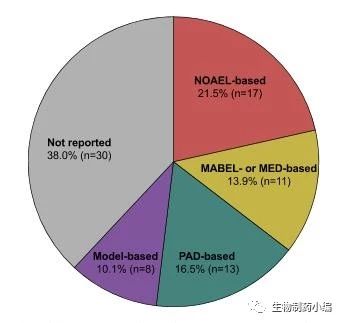
根据这项研究,基于NOAEL的方法仍然是mAbs进行FIH研究的最常用的确定人体起始剂量的方法,而基于模型的方法的使用则少得多。超过三分之一的FIH研究采用NOAEL的方法,这是使用模型方法的两倍。下面将简明扼要地介绍如何使用NOAEL方法以确定单克隆抗体药物在首次人体临床研究时的起始剂量。
如何使用基于NOAEL的方法计算MRSD
FDA行业指南«估算成人健康志愿者中新药首次临床研究的最大安全起始剂量»描述了这个计算过程与方法,用于推导在健康成人志愿者中新分子实体进行首次人体临床试验的最大推荐起始剂量(MRSD),并将这个过程标准化, 其目的是确保志愿受试者的安全。
在FIH研究中, 应该避免初始临床剂量导致毒性, 同时应该选择允许合理快速达到临床I期的研究目标(例如,评估治疗剂的耐受性,药效学或药代动力学特征)的剂量。在确定MRSD时,应考虑所有相关的临床前数据,包括药理活性剂量的信息,完整的化合物毒理学特征以及其药代动力学(吸收,分布,代谢和排泄)。从低于MRSD的剂量开始始终也是一个选择,可以满足一些特别临床试验目标。
推算MRSD的过程应在分析所有毒理数据后开始。虽然只有NOAEL直接用于计算MRSD,但其他数据(暴露/毒性关系,药理学数据或先前的相关药物临床经验)可能会影响选择最合适的物种,种属縮放和安全系数。首先, 确定每种被研究物种的NOAEL,然后使用适当的比例因子将其转换成HED。对于大多数有生物体系统暴露的药物,这种转化应基于剂量对于体表表面积的标准化。在没有进一步可用的信息时,体表面积换算是近似等效暴露的标准方法。体表面积标准化和动物剂量至人体剂量的比例外推应该通过将所研究的每种动物物种的NOAEL除以适当的体表面积换算系数(BSA-CF)在一个步骤中完成。这个换算系数是一个无单位的数字,它将每种动物物种的mg / kg剂量转换为人体的mg / kg剂量。该人体剂量相当于以mg / m2计算的动物的NOAEL,被称之为人体等效剂量(HED)。产生最低HED的物种被称为最敏感的物种。
如果有数据表明某特定物种与评估人类风险更相关而是最适合的物种时,该物种的HED可用于随后的计算,无论该物种是否最敏感。这种情况似乎特别适用于生物药,其具有与人类靶点蛋白结合的高选择性以及对常用于毒性研究的物种靶点的有限亲和性。在这种情况下,应该进行体外与靶点结合和功能的研究,以便在设计毒性研究之前选择适当的相关物种。请参阅ICH工业S6临床前安全性评估生物技术衍生药物指南以获得更多细节。但是,如果在不太相关的动物物种中观察到严重的毒性,那么在确定用于计算HED的物种时也应考虑这些毒性。
之后,应该对HED应用一个安全系数,以确保人体上第一次给药不会造成不良影响。安全系数的使用是考虑到人类对动物模型所预测的毒性作用可能更敏感,生物利用度可能因物种不同而不同,并且所研究的动物模型也不能评估所有人体上可能的毒性。一般来说,应该使用至少10倍的安全系数。将HED除以安全系数即获得MRSD。在动物研究中的安全问题或设计缺陷可能需要增加安全系数,从而进一步降低MRSD。另一方面,对具有广泛人类临床前和临床经验的,特征良好的药物类别可以降低默认的安全系数。当然,低于MRSD的剂量也可用作首次临床研究实际起始剂量,但这不是我们的主要目标。 MRSD以mg / kg为单位,这是用于临床I期的常用给药的计算方式。以下进一步阐明有关的4个步骤。
第1步:未观察到不良反应水平(NOAEL)的确定
确定MRSD的第一步是评估可用的动物数据,以确定NOAEL。在选择人体起始剂量时,应使用以下NOAEL的定义:最高剂量水平与对照组相比不会显著增加不良反应。在确定NOAEL时,应考虑生物学意义上的不良反应,即使它们不具有统计学意义。从适当的动物研究中得出的NOAEL是普遍接受的安全性的基准,也是确定健康(或无症状)人体中新药的相对安全的起始剂量的出发点。
在非临床毒理学研究中,基本上有三种类型的结果可用于确定NOAEL:(1)明显的毒性(例如临床症状,宏观和微观病变); (2)毒性的替代标记(例如血清肝酶水平); 和(3)夸大的药效作用。尽管不良作用的性质和程度可能因不同类型的药物而有很大差异,并且在许多情况下,专家们会对不良反应的评估也不一致;但作为一般规则,该不良作用是指由药物的初始剂量在成人健康志愿者中导致的,不可接受的不良作用。
第2步:基于体表面积的人体等量剂量转换
在相关动物研究中的NOAEL被确定之后,它们将被转换成HED。通常假定,动物全身性使用的药物的毒性终点与药物剂量(标准化为mg / m2)之间的关系在物种间可以比例外推。同时,基于需要保守的起始剂量估计以增加临床试验安全性的考虑,一般也应当选择基于体表面积的校正因子将NOAEL剂量转化为HED,以得到最终的FIH起始剂量, MRSD。从NOAEL到HED的转换系数见表1。
表1。转换动物剂量为人体等效剂量的转换系数(依体表面积法,摘自FDA指南)。
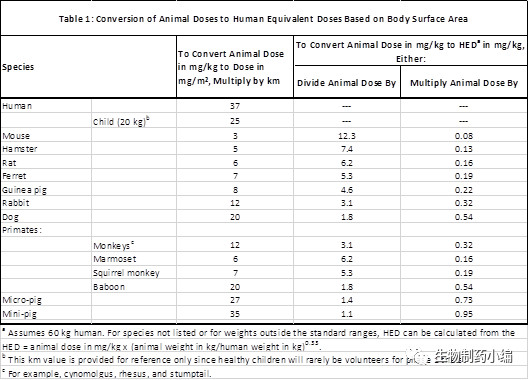
表1中将动物NOAEL转换为HED的转换因子的推导是基于如下假设:即当将剂量标准化为体表面积时,NOAEL与HED是1:1的比例。 然而,有时基于体重的缩放方法,即设定HED(mg/kg)= NOAEL(mg/kg),可能更合适。这样得出的HED(mg / kg)数值,相对于小鼠,大鼠和狗,分别是默认的mg/m2方法的12倍,6倍和2倍。如果考虑使用mg/ kg缩放比例,须有数据表明NOAEL(mg / kg)在所有研究过的物种间的数值相近。否则应该遵循基于mg/m2的缩放方法而得出更安全的MRSD。此外,FDA特别指出对于血管内注射的分子量大于100,000道尔顿的蛋白质药物,建议使用基于mg/kg的比例缩放。详情请阅FDA指南。
第3步:最适宜的动物物种选择
从所有动物毒理学研究中得出的NOAELs中确定HEDs之后,下一步是选择一个HED用于随后推导MRSD。该HED应该从最合适的物种中选择。在缺乏明确的物种相关性数据的情况下,用于推导成人健康志愿者的MRSD的最合适物种应默认是最敏感的物种,即可判定的拥有最低HED的物种。
如果不采用默认的选择,可能影响最适合物种选择的因素通常是:
(1)不同物种间药物的吸收,分布,代谢和排泄(ADME)相当地不同。当确定新药的MRSD时,人体的吸收,分布和消除参数仍是未知。然而,体外研究得出的比较代谢数据是有价值的。这些数据结合在多种动物体内的代谢物特征,相对于人体体外数据,在解读动物种属间存在的HEDs的明显差异时特别相关。
(2)药物类别经验可能表明某个特定动物模型的更能预测人体的毒性。选择某些生物药(例如人类蛋白质)的最合适物种需要考虑这些药物所特有的各种性质,诸如该动物物种是否表达相关受体或表位。这肯定会影响物种选择(参见关于生物技术衍生制剂的临床前安全评估的ICHS6指南)。类别经验表示以前的研究表明,某个特定的动物模型更适合于评估某类特定药物的安全性。
第4步:应用安全系数
一旦确定了最适当物种的NOAEL的HED,就应该使用安全系数来为接受初始临床剂量的人类受试者提供安全边界。这个安全系数考虑到从动物毒性研究到人体研究的比例外推时的可变因素:(1)由于人体与动物药理活性敏感性增加而引起的不确定性; (2)探测动物某些毒性的困难(例如,头痛,肌痛,精神障碍); (3)药物受体密度或亲和力的差异; (4)预想不到的毒性; 和(5)药物ADME的种属间差异。所有这些可变因素可以通过降低HED剂量来适应。
将HED除以安全系数即确定FIH临床研究的MRSD。默认的安全系数为10. 这是历史上接受的数值。当有理由增加关注时应该提高安全系数;在有可用数据提供额外的安全保证时,可以降低安全系数。这样的增加或减少程度主要是一个判断的问题。评估者有责任在与默认值10不同时,特别是如果它低于10,明确地解释其背后的推理。
目前,不同的人体起始剂量确定方法使用的安全因子差异很大。基于MABEL或MED的方法比其他人体起始剂量测定方法的安全因子中值(median value)要小得多。使用安全因子是考虑到种属间潜在的差异,等不确定因素,可作为确保FIH研究中的首次剂量在人体不会发生毒性的一个额外手段。因此,较小的安全因子表明对该药物在人体上的安全性的有更大信心。与NOAEL和其它方法相比,MABEL方法比其他方法总是得出更小的HED。因此,基于MABEL的安全因子往往比其他方法小。
总而言之,NOAEL方法的剂量选择是基于最小的毒性风险,而不是选择对人体具有最小药理活性的药物剂量。
确定MRSD的假设性例子
NOAEL方法应用体表面积的指数(0.67),这代表了种属间代谢速率的差异,可以在动物和人类之间转换剂量。因此,HED由以下等式确定:
HED(mg/kg) = Animal NOAEL (mg/kg) × (BWanimal[kg]/BWhuman[kg]) (1–0.67) (1)
例如,对于某新药物,重约150g的大鼠的NOAEL值为18 mg/kg。要计算人体研究的起始剂量,使用公式(1)得出:
HED(mg / kg) = 18 × (0.15 / 60)0.33=18 x 0.14 = 2.5 mg / kg
因此,对于60公斤的人,剂量为60×2.5 mg/kg = 150 mg。该HED值除以安全系数10得出MRSD为15 mg。
确定MRSD的真实例子
Janssen R&D的CNTO 5825是抑制人IL-13与IL-13Ra1和IL-13Ra2结合的人源抗白介素-13(IL-13)单克隆抗体。在CNTO5825的4周GLP毒性研究中,NOAEL在大鼠和食蟹猴中均为100mg / kg(Janssen R&D未发表的数据)。对大鼠和食蟹猴的4周GLP毒性研究中的CNTO 5825的NOAEL通过乘以0.16的大鼠因子和食蟹猴的0.32因子(表1)转化为人体等效剂量(HED):
大鼠:HED(mg /kg) = 100 mg/kg × 0.16= 16 mg/kg
食蟹猴:HED (mg /kg) = 100 mg/kg × 0.32=32 mg/kg
因此,基于大鼠NOAEL的 HED为16 mg / kg,MRSD为1.6 mg / kg;基于食蟹猴NOAEL的HED为32 mg / kg,MRSD为3.2 mg / kg。所以,大鼠是最敏感物种。将至少10倍安全系数应用于HED可以获得FIH最大推荐起始剂量(MRSD)。由于CNTO5825对大鼠或食蟹猴IL-13的结合亲和力比对人的结合亲和力低7倍至15倍,故对大鼠的HED施加额外的安全系数160以获得CNTO 5825在FIH的MRSD为0.1 mg / kg。
注意:本例中转换因子的数值与上例中略有不同,主要是使用动物和人体体重不同引起的。
种属缩放指数对计算HED的影响
FDA在其指南中进行分析以确定了种属缩放指数对动物剂量转化为HED的影响。根据Dose (mg) = aBWb 的指数关系,其中BW为体重, b为种属縮放指数,可以推导出以下用于将动物NOAEL剂量转化为HED的等式:
HED= animal NOAEL x (BWanimal/BWhuman)(1-b) (2)
通常,对于mg / m2的标准化指数b为0.67,但是一些研究(包括最初的Freireich数据)表明,当b = 0.75时,最大耐受剂量(MTD)在物种间的比例外推最佳。 FDA的机构间药代动力学小组也建议在致癌性研究中使用BW0.75在动物种属间来比例外推剂量(EPA 1992)。然而,并无数据支持将NOAEL转化为HED的最佳方法。因此,下面使用(BWanimal/ BWhuman)0.33或(BWanimal / BWhuman)0.25在一系列动物上计算转换因子,以评估使用b = 0.75或 0.67对选择起始剂量的影响。表2显示了计算结果。使用0.75的种属缩放指数对较小物种小鼠和大鼠的转换因子有很大影响。当然,小鼠并不常用于支持首次人体临床试验的毒理学研究。
表2:种属缩放指数对转换因子的影响。
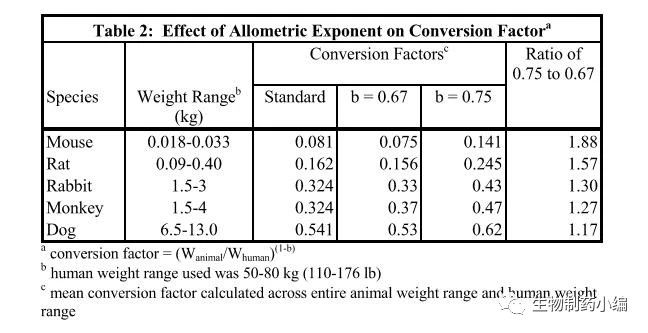
此外,有数据表明,当剂量按mg/m2标准化时,大鼠和人体内血浆浓度对时间曲线下的面积(AUC,即暴露量)相关性良好(Contrera et al。1995)。 FDA的最终意见是应该保持基于体表面积校正因子(即b = 0.67)将NOAEL剂量转换为HED的方法,来选择健康志愿者初始研究的起始剂量,因为:(1)mg/m2表准化广泛地用于整个毒理学和药代动力学研究界; (2)mg/m2标准化提供更保守的转换; (3)没有数据可以支持用于转化NOAEL的更优方法; (4)该转换计算容易,并且CDER在建立以mg/m2为基础的安全起始剂量方面有着丰富的经验。显然,采用b = 0.67 而不是b = 0.75是基于经验,受试者安全和惯例的考虑,而不是基于纯科学的考虑。但从安全和便于实际操作的角度出发,这也是可以理解和接受的。
总结和前瞻
本文简明扼要地介绍如何使用NOAEL方法以确定单克隆抗体药物在首次人体临床研究时的起始剂量。读者可从参考文献,特别是FDA的相关指南中,获取有关NOAEL方法的详细和权威性的阐述。
NOAEL方法使用在小分子药物有着良好的安全记录; 而且易懂易学,使用方便。可以任意选择的安全系数可以使得这种方法非常保守。其不足是忽视了在研药物在生物种属间药理学上的差异,如结合亲和力和效力。
尽管基于NOAEL的方法仍然可能是最常用的方法,但预计未来将基于MABEL的方法在使用单克隆抗体的FIH研究中将会更频繁地使用。基于MABEL的方法似乎比其他的人体起始剂量确定方法能更有效率地实现FIH临床实验的目标。小编将在本系列的下一篇文章中描述MABEL方法。
特别声明
本文如有疏漏和误读现有数据的地方,请读者评论和指正。所有引用的原始信息和资料均来自已经发表学术期刊,官方网络报道, 等公开渠道, 不涉及任何保密信息。参考文献的选择考虑到多样化但也不可能完备。欢迎读者提供有价值的文献及其评估。
扩展阅读
简化种属縮放定律用于预测治疗性单抗的人体药代动力学 2018-03-13 游方和尚生物制药小编
参考文献
1.US Food Drug Administration.Guidance for Industry: Estimating the Maximum Safe Starting Dose in InitialClinical Trials for Therapeutics in Adult Healthy Volunteers. Rockville, MD:Food and Drug Administration; 2005. [Accessed July 6, 2005]. Available from: http://www.fda.gov/downloads/drugs/guidances/ucm078932.pdf.
2.Committee for Medicinal Products for Human Use. Guideline on Strategies to Identify and Mitigate Risksfor First-in-Human Clinical Trials with Investigational Medical Products.London, UK: European Medicines Agency; 2007. [Accessed July 19, 2007].Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500002988.pdf.
3.Ivy SP, Siu LL, Garrett-MayerE, Rubinstein L. Approaches to phase 1 clinical trial design focused on safety,efficiency, and selected patient populations: a report from the clinical trialdesign task force of the national cancer institute investigational drugsteering committee. Clin Cancer Res. 2010;16(6):1726–1736
4.Suh HY,et al, Determination of the starting dose in thefirst-in-human clinical trials with monoclonal antibodies: a systematic review of papers published between 1990 and 2013 . Drug Des Devel Ther. 2016; 10:4005–4016
5.Wang W, Wang E, Balthasar J. Monoclonal antibody pharmacokinetics and pharmacodynamics. Clin Pharmacol Ther. 2008; 84 (5):548–558.
6.Zhao L, Ren T-h, Wang DD.Clinical pharmacology considerations in biologics development. Acta PharmacolSin. 2012; 33 (11):1339–1347.
7.Nnane IP et al, Non-Clinical Pharmacokinetics,Prediction of Human Pharmacokinetics and First-in-Human Dose Selection for CNTO5825, an Anti-Interleukin-13 Monoclonal Antibody.Basic Clin Pharmacol Toxicol.2015; 117(4):219-25. doi: 10.1111/bcpt.12391.
8.Nair AB, et al, A simple practice guide for dose conversion between animals and human. J Basic Clin Pharm. March 2016-May 2016; 7(2): 27–31. doi:10.4103/0976-0105.177703
9.EPA, 1992, A Cross-Species Scaling Factor for Carcinogen Risk Assessment Based on Equivalence of Mg/Kg0.75 /Day,Federal Register, 57:24152-24173.
10.Freireich, EJ, et al 1966, Quantitative Comparison of Toxicity of Anticancer Agents in Mouse, Rat, Hamster, Dog, Monkey, and Man,Cancer Chemotherapy Reports, 50:219-244.
11.Mordenti, J, 1986, Man Vs. Beast: Pharmacokinetic Scaling in Mammals, Journalof Pharmaceutical Sciences, 75:1028-1040.
12.Reigner, BG and KS Blesch, 2002, Estimating the Starting Dose for Entry into Humans: Principles and Practice, European Journal of Clinical Pharmacology,57:835-845.
13.Schein, PS, RD Davis, S Carter, J Newman, DRSchein, and DP Rall, 1970, The Evaluation of Anticancer Drugs in Dogs andMonkeys for the Prediction of Qualitative Toxicities in Man, Clinical Pharmacology and Therapeutics, 11:3-40. Spector, WS (ed.), 1956, Handbook of Biological Data, pp. 175, Philadelphia:W.B. Saunders Co.
14. Contrera, JF, AC Jacobs, RP Hullahalli, M Mehta, WJSchmidt, and JA DeGeorge, 1995, Systemic Exposure — Based Alternative to the Maximum Tolerated Dose for Carcinogenicity Studies of Human Therapeutics,Journal of American College of Toxicology, 14:1-10.
15.Travis, CC and RK White, 1988, Interspecies Scalingof Toxicity Data, Risk Analysis, 8:119- 125.
16.Watanabe, K, FY Bois, and L Zeise, 1992,Interspecies Extrapolation: A Reexamination of Acute Toxicity Data, Risk Analysis, 12:301-310.


