


编者按
Emicizumab是在研双特异性抗体项目中进展最快的一个,有望成为第三个上市的双特异性抗体和血友病治疗领域第一个抗体。小编在上一篇文章中解读了血友病治疗领域远未满足的临床需求和Emicizumab巨大的市场潜力。作为上一期文章的续篇,小编将在本文中为读者解读Emicizumab成功背后的研发历程。
一、简介
全球目前已经上市了费森尤斯公司(Fresenius)的Removab (Catumaxomab,2009)和安进的Blincyto(Blinatumomab,2014)两个双特异性抗体药物。Emicizumab通过模拟FVIIIa的辅因子功能,增强FIXa和FX的相互作用并增大FIXa催化FX形成FXa的催化速率常数,最终代替FVIIIa在凝血级联反应中的作用并治疗A型血友病,有望成为第三个上市的双特异性抗体。
Catumaxomab的技术平台为Triomab,但因其具有FcγR共激活作用而不能采用系统性静脉注射给药,以及由于其为鼠科抗体具有极高的免疫原性而不能重复给药限制了其使用。Blincyto的技术平台为BiTE,但因其不具备天然抗体的Fc结构而平均半衰期仅为2.11小时,一定程度上限制了其应用。
除以上有实际应用案例的2个双特异性抗体技术平台以外,目前在研双特异性抗体项目的技术平台超过20个,如Tandem scFV、IgG-scFV、DVD-Ig、CrossMab和CLC-IgG等。

中外制药的Emicizumab选择了非对称性IgG形式的双特异性抗体,具有以下3个特点:(1)两个臂分别特异性识别FIXa和FX;(2)具有较长的半衰期;(3)具有天然形式的IgG结构。
二、临床前开发
2.1 分子设计和优化
如果不对抗体的结构做任何改造和优化,在一个细胞中同时表达两条不同轻链和两条不同重链可组成10种抗体分子,其中4种为重链和轻链均相同的抗体(每种含量为6.25%)而另外6种为重链和轻链至少有一不同的抗体(每种含量为12.5%),因此目标双特异性抗体仅占12.5%,这会给双特异性抗体的工业化生产带来极大的挑战。

中外制药采用“Knobs-into-Holes(KiH)”、“Common Light Chain-IgG(CLC-IgG)”和“Multidimensional Optimization(多维优化)”等技术,分别对双特异性抗体的Fc、LC和可变区进行分子工程改造和优化得到了Emicizumab,克服了前两个上市双特异性抗体在工业化生产和规模化应用方面的局限。
2.1.1 Fc设计
Emicizumab的Fc设计采用了Genentech在1990年代后期发明的KiH技术,在一条重链的CH3区域引入一个较大的氨基酸残基(如酪氨酸)形成“knob”而在配对的另一条重链CH3的相应区域引入一个较小的氨基酸残基(如丙氨酸、甘氨酸等)形成一个“hole”,这样两条不同的重链就可通过空间互补形成异源二聚体。
通过KiH技术可使重链异源二聚体增加,但仍然存在knob-knob和hole-hole形式的重链同源二聚体,knob-knob重链同源二聚体比例较少,而hole-hole重链同源二聚体含量较多。为了增加重链异源二聚体的比重和稳定性,进一步通过对重链CH3区域的相应氨基酸突变为半胱氨酸,形成重链异源二聚体间二硫键(S-S),进一步增加重链异源二聚体的比例。

除此之外,通过中外制药科学家Tomoyuki Igawa等在2006年发明的“Electrostatic Steering(静电转向)”技术在两条不同重链的CH3区域分别引入带负电和带正电的氨基酸残基,使重链同源二聚体由于静电排斥而减少而重链异源二聚体由于静电吸引而增加。
经过KiH、S-S和Electrostatic Steering三种技术对Fc的CH3区域进行改造后,重链异源二聚体(不考虑轻链的配对形式)的比例可达90%以上。
2.1.2 LC设计
重链异源二聚体含有四种不同形式的轻链配对,因此具有正确配对轻链的重链异源二聚体比例仅占25%。同时,经过Fc的CH3区域改造可使重链异源二聚体比例达到90%以上,因此在表达产物中目标双特异性抗体比例为90%×25%=22.5%以上。这一数字虽然较最初的12.5%有一定提升,但仍然难以满足工业化生产和应用的需求。

为了解决轻链不同造成的正确配对双特异性抗体比例低的问题,中外制药发明了CLC-IgG技术。CLC-IgG使两条具有不同特异性的轻链改造为具有不同特异性的一条共同轻链。分别用人的FIXa和FX免疫动物,得到各约200个抗体,分别将其VL和VH与人源轻链恒定区 κ/λ和IgG2/IgG4进行融合,然后具有不同特异性的重链进行组合得到约40000个双特异性抗体。
Emicizumab是一种模拟FVIIIa的催化辅因子功能的抗体,其活性不仅仅需要结合FIXa和FX形成空间构象合适的复合物将FX催化为FXa,而且催化结束后还需要快速释放FXa发挥后续的凝血级联反应并持续进行“结合-催化-释放”过程。对Emicizumab来说并非亲和能越强越好,尤其是对于FX来说(FVIIIa结合FX的Kd=1-3 μM),因此不能用展示技术等通过亲和力强弱进行筛选的平台进行筛选。基于以上原因,只能通过催化反应测定是否具有FVIIIa活性进行筛选。通过催化活性测定,从重组表达得到的约40000个双特异性抗体中筛选得到了94个具有FVIIIa活性的双特异性抗体(耗时费劲的体力活!)。

94个产生FVIIIa活性的双特异性抗体具有188条不同的轻链,因此可以组合形成188个具有相同轻链而重链不同的抗体。通过活性筛选得到了活性最高的具有共同轻链的双特异性抗体c1。为了得到活性更高的双特异性抗体,其轻链c1L与另一重链同源的轻链c2L具有>85%同源性的轻链c3L(由于轻链c2L替换c1L后没有活性,因此不可用)进行FR/CDR shuffling得到了具有最高活性含共同轻链的双特异性抗体BS15L,之后对其FR区进行人源化改造得到hBS15L(hBS1)。
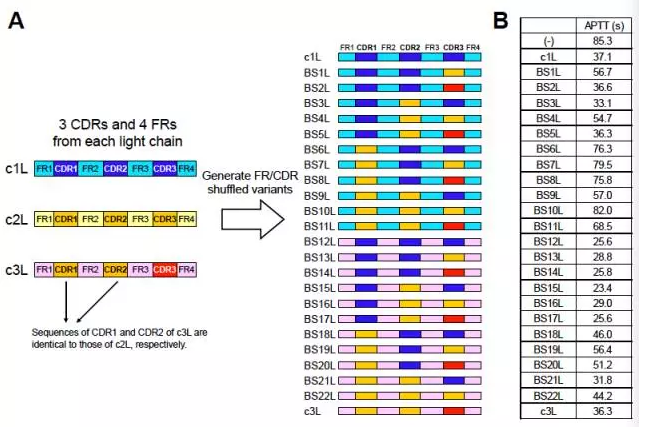
经过CLC-IgG改造,重链异源二聚体的轻链配对形式由4种可能变为1种,因此具有正确配对轻链的重链异源二聚体比例由25%变为100%。同时,如前所述经过Fc的CH3区域改造可使重链异源二聚体比例达到90%以上,因此在表达产物中目标抗体比例为90%×100%=90%以上。
通过KiH和CLC-IgG改造,特别是CLC-IgG改造,可极大的提高表达产物中目标产物的含量(由12.5%提高为>90%),大大提高了表达量并减轻了下游纯化的压力。
2.1.3 可变区优化
得到人源化具有共同轻链的双特异性抗体hBS1后,中外制药通过CDR区突变,对其活性、PK、pI、溶解性、稳定性和免疫原性等进行了优化,得到了各项指标满足生产和应用的双特异性抗体hBS910。

第一步:对hBS1的CDR区进行突变,得到活性大幅提升的hBS106,然而其PK参数与天然IgG4存在较大差距。
第二步:通过hBS106的同源模型解析发现anti-FIXa的Fv表面正电簇影响了其PK,通过多种尝试,对轻链的突变Tyr30Glu可在不降低活性的情况下中和正电簇改善PK得到hBS128。
第三步:虽然表达产物中目标抗体的含量达90%以上,但还是存在一定量与目标抗体理化性质极为相似的同源二聚体(多为hole-hole形式)。同源二聚体的存在,一方面为工业化生产的下游纯化带来困难,另一方面带入最终产品后会与目标抗体竞争性结合FIXa和FX进而影响其活性和应用。中外制药采用了pI工程理念对重链的可变区进行突变增加目标抗体hBS366/hBS376和同源二聚体的pI差异,方便下游采用离子交换色谱去除同源二聚体。
第四步:皮下给药(体积<1.5 mL)一般要求目标抗体具有较高的浓度(如100 mg/mL)。然而hBS376在较低浓度下就出现了分相及沉淀现象。通过将可变区的疏水氨基酸突变为亲水氨基酸,得到了溶解度大幅提升(>100 mg/mL)的hBS560。
第五步:通过hBS560加速稳定性试验发现了HCDR3区的Asn99存在脱酰胺现象,而且导致活性降低。但仅突变Asn99影响其活性,最终找到了去除脱酰胺的突变组合His98Arg 和 Asn99Glu得到hBS660。
第六步:去除双特异性抗体潜在的T细胞表位,通过Epibase(Lonza)和EpiMatrix(EpiVax)免疫原性预测系统预测,得到了免疫原性与曲妥珠单抗相似的双特异性抗体hBS910。

通过KiH、CLC-IgG和多维优化等技术,分别对双特异性抗体的Fc、LC和可变区进行分子工程改造得到了具有应用潜力的hBS910(ACE910,Emicizumab,至少筛选了910个双特异性抗体!)。经过TGA(凝血酶生成)试验测定,针对FIXa、FX的单抗以及两种抗体混合物均不具备FVIIIa的催化活性,而双特异性抗体Emicizumab的活性可与rhFVIII比拟,并且不受FVIII抑制物的影响,显示出其潜在的临床应用价值。
2.2 非临床评价
A型血友病的动物模型有鼠、狗、兔、羊和猪等,但跟人的种属差异性较大,因此中外制药采用了非人灵长类动物食蟹猴作为双特异性抗体的非临床评价动物模型。由于不存在先天性的A型血友病食蟹猴,在开发过程中中外制药通过向食蟹猴注射FVIII抑制物的方式构建了获得性A型血友病食蟹猴,以评价双特异性抗体的凝血活性。

通过三项获得性A型血友病食蟹猴的动物试验,证明了Emicizumab可预防肌肉、皮下组织及关节出血,并且具有较长的半衰期(可达20天),而且可以长间隔皮下给药(皮下注射生物利用度约为100%)。
进一步的人体临床研究证明了其比重组和血浆来源FVIII在预防A型血友病患者出血方面具有更优越的安全性、有效性和患者顺应性(可每周皮下给药一次,正在计划启动每月皮下给药一次的临床试验)。
由此可见,Emicizumab的初步成功的前提是中外制药进行了充分的临床前研究,克服大量在双特异性抗体分子设计、先导抗体筛选、双特异性抗体理化性质优化和动物模型构建等过程中遇到的难题。从中外制药申请的相关专利看,该项目可能开始于2004年以前,花了至少6-7年时间解决在双特异性抗体分子设计、先导抗体筛选构建和工业化生产等方面的难题,直至2012年8月才进入I期临床试验。
三、Baxalta专利诉讼
作为血液制品行业外来者的中外制药(主要为抗肿瘤药)取得了近20年来血友病治疗领域最大的突破,那么传统血液制品巨头有何反应呢?也许有一件事值得关注:Baxalta在2016年5月6日对中外制药发起专利诉讼,指控Emicizumab侵犯了其专利JP4313531。中外制药也在2016年5月11日发表声明,称其会收集证据证明Emicizumab没有侵犯JP4313531专利。
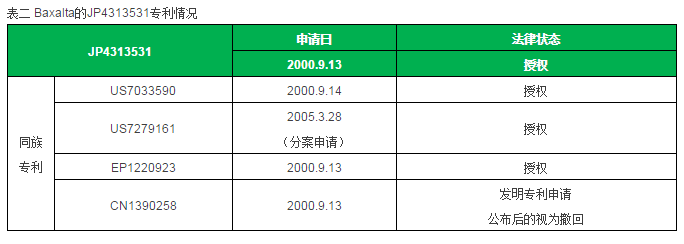
Baxalta的JP4313531的申请日为2000.9.13,其同族专利在美国、欧盟都已经获得授权,但在中国未获得授权(“发明专利申请公布后的视为撤回”),美国专利在2005.3.28申请了分案专利,也获得了授权。该系列专利申明的权利为特异性结合FIX或FIXa的抗体和抗体衍生物(含双特异性抗体)的制造方法、可变区的序列及其用途,但是通篇没有看见双特异性抗体的具体实施例。
Emicizumab的核心专利为JP5246905,其同族专利在美国和中国都已获得授权,在欧盟还处于审查阶段,申请日都为2011.11.17。该专利申明的权利为特异性结合FIXa和FX的双特异性抗体的一系列氨基酸序列及其用途。
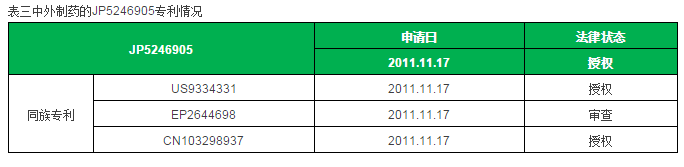
如果判决中外制药侵权,Emicizumab在日本、欧洲可能会延迟至2020年才能上市,在美国更是会延迟至2025年上市,而在中国不受影响。考虑到Emicizumab目前处于III期临床,中外制药计划在2017年提交NDA,一切顺利有望在2018年获批,因此一旦被判侵权会严重影响Emicizumab在主要用药国家的上市时间。
如果判决中外制药不侵权,Emicizumab一旦上市,会显著减少现有A型血友病药物的使用量,对于Baxalta已经上市或在研的A型血友病药物将是巨大的打击。
此次专利诉讼也许仅是双方专利战的开端,可能还会在其他国家和地区上演类似的剧情。对于患者来说,由于专利问题延迟具有优秀临床价值的药物上市也许不是一个好事。不管采用何种解决方案,希望各国监管方、法院和企业等参与者不要让专利问题成为优秀药物成为上市的绊脚石。
血友病是关注度较高的罕见遗传疾病,治疗药物不断革新,新药研发异常激烈。基因疗法也许是解决遗传疾病的最终手段,2016年7月21日Spark Therapeutics和辉瑞公司合作进行的B型血友病治疗项目SPK-9001被FDA授予突破性疗法认证。
参考文献
1. Emicizumab.Thomson Reuters.
2. Chugai Pharmaceutical官网.
3. Spark Therapeutics官网.
4. ‘Knobs-into-holes’ engineering of antibody CH3 domains for heavy chain heterodimerization(1996).
5. An efficient route to human bispecific IgG(1998).
6. A bispecific antibody to factors IXa and X restoresfactor VIII hemostatic activity in a hemophilia A model(2012).
7. Anti-factor IXa X bispecific antibody ACE910 prevents joint bleeds in a long-term primate model of acquired hemophilia A(2014).
8. Anti-factorIXa X bispecific antibody (ACE910) hemostatic potency against ongoing bleeds in a hemophiliaA model and the possibility of routine supplementation(2014).
9. engineering the variable region of therapeutic IgGantibodies(2011).
10. Identification and multidimensional optimization ofan asymmetric bispecific IgG antibody Mimicking the Function of Factor VIII Cofactor Activity(2013).
11. Progress in overcoming the chain association issuein bispecific heterodimeric IgG antibodies(2012).


