

摘要:近些年来,治疗性抗体及抗体类蛋白已经成为欧美新批准药物的一大组成部分,此类药物的临床试验数量呈迅速增长的趋势。一个可成功开发成商业化药物的治疗性蛋白,不仅应具有理想的药效、安全性和药代动力学特性,还应具有理想的理化特性,使得其稳定性能够满足生产、制剂工艺的技术要求。这一系列理化特性的评价,也称为“成药性”或“可生产性”评价。本文总结了目前成药性评价方法的研究进展。
关键词:治疗性蛋白、成药性、可生产性、理化性质、稳定性、制剂
Developability Assessment of Therapeutic Proteins
Abstract: In the last decades, therapeutic antibodies and antibody-like molecules constitute a big class of newly approved drug in western countries. Currently, clinical trials of such drugs are still growing fast. A therapeutic protein which can be successfully commercialized, should not only exhibits desired efficacy, safety and pharmacokinetics, but also achieves drug-like physiochemical properties to ensure the stability meets technical requirements of manufacturing and formulation process. Methods of so called “developability” or “manufacturability” assessment are summarized in this paper.
Key word: therapeutic protein;developability; manufacturability; physiochemical properties; stability; formulation
近年来,基于单克隆抗体的治疗性药物成为了制药企业研发管线中最重要的一部分。据统计,2016年处于临床研究中的抗体类药物分子数量在已超过了470个[1],适应症范围覆盖了肿瘤、自身免疫、眼科及一些罕见病等多个方面。大分子蛋白药物在原液、制剂生产,及临床给药时常遇到的一个问题是蛋白的不稳定性。一方面由于蛋白质天然的稳定性显著低于小分子化药,另一方面为了达到天然蛋白所不具有的治疗特性,研究者们还应用蛋白质工程设计出了各种非天然蛋白,如双特异性抗体、融合蛋白等。而同时这些非天然蛋白的稳定性常常更加成为问题。一些理化特性较差的蛋白常常在生产、储存、给药过程中出现研究者不想看到的化学修饰、断裂和聚集等现象,这大大影响了药物的产率、活性,高分子聚集体还会造成免疫原性等方面的安全性问题。
过去很多研究机构主要基于生物学活性来筛选候选分子,其可生产性的相关影响因素在分子发现阶段并未纳入考量范围。但这些分子常常在推进到后期生产工艺开发阶段时,遇到稳定性等技术方面的问题而无法顺利商业化,从而导致大量资源被浪费。近5年来,越来越多的研发机构开始将成药性评价也纳入蛋白药物发现阶段,以得到最佳的药物分子。与小分子药物已有简单成熟的成药性评价标准:“里宾斯基五规则”[2]不同,大分子药物的成药性评价目前尚无类似的评价标准。本文结合近年来各方面的研究进展,将所报道的各种大分子成药性评价方法进行了综述。
药物发现的早期阶段一般采用计算机虚拟计算的方式对蛋白序列进行初步的评估,包括蛋白电荷性质的计算,潜在翻译后修饰位点,及可能的化学降解位点的寻找。如发现候选分子上有潜在的不稳定位点,可进一步使用少量纯化的样品进行强制降解实验来进行序列比较,并对降解位点进行鉴定。根据虚拟和实际实验结果,选择潜在不稳定位点较少的序列,或对高风险氨基酸位点进行突变,从而避免后期制造过程中这些风险位点造成的失败。
计算机基于氨基酸序列计算出的蛋白理论等电点(pI)和实际值有时会有一些差距,例如结构上相近的氨基酸侧链可能形成盐桥,进而影响其pKa值。罗氏的研究人员对比了9个单抗的理论预测pI和实验测得pI,相差约为± 1 [3]。因此,在用于成药性评价时,基于序列的理论预测准确度也已经足够。抗体类分子一般要求等电点高于7,从而保证该分子能够适用于下游纯化工艺平台中的阴、阳离子交换层析。另外,蛋白只有处在远离等电点的pH下,才能保证相对合适的溶解度,并控制聚集。因此候选分子的等电点也应远离生理条件的pH。
除了等电点,候选分子的整体电荷分布也应进行考量。极端不对称的表面电荷分布与蛋白自身相互作用、聚集和粘度等有关。由于表面电荷分布与蛋白高级结构有关,进一步的叙述见下文。
糖基化(N-Glycosylation):抗体可变区糖基化的存在可能影响其结合活性和药代动力学特性,也是产品异质性的一个来源。N-糖基化固定发生在Asn-X-Ser/Thr位点,X为除脯氨酸之外的任意氨基酸,因此基于蛋白氨基酸序列的一级结构,即可分析推测出潜在的N-糖基化位点。尽管并不是所有这些位点在生产时均会被糖基化,但后期工艺的改变仍然可能改变这些位点的暴露状态,继而改变糖基化状态,带来产品特性改变的风险。
游离巯基(Unpaired cysteine):候选抗体可变区的半胱氨酸可以通过序列搜索而发现。未配对的半胱氨酸残基可结合细胞培养基中的游离半胱氨酸、谷胱甘肽,及其他含有游离巯基的化合物,带来产品异质性。更糟糕的是,游离巯基在一些条件下(如pH > 7),可能会形成二硫键错配,并可能造成二硫键介导的二聚体甚至多聚体的产生。
糖化位点(Glycation):赖氨酸的糖化作用是指氨基酸序列中的赖氨酸残基侧链与还原性糖,如葡萄糖、果糖发生的美拉德反应(Maillard reaction)[4]。该反应也是产品异质性的另一个来源,主要发生在细胞培养、储存等过程中,以及稀释至葡萄糖注射液中用于临床静脉滴注时。糖化作用在抗体中较为常见,并没有特异性的作用基序。在所报道的糖化发生案例中,有一些会影响抗体活性,而另一些则不会,这可能与其发生的具体位置有关。因此糖化作用仍然是成药性评价中值得考虑的一个因素,如果在候选抗体分子CDR区发现有赖氨酸,则应慎重考虑可能的糖化作用及其影响。在实际样品测试中,由于美拉德反应会带来颜色的褐变,可用于推测溶液中蛋白糖化作用的发生[5];分离糖化和非糖化的蛋白可采用硼酸亲和层析或离子交换层析;质谱法则可用于糖化位点的最终确认。
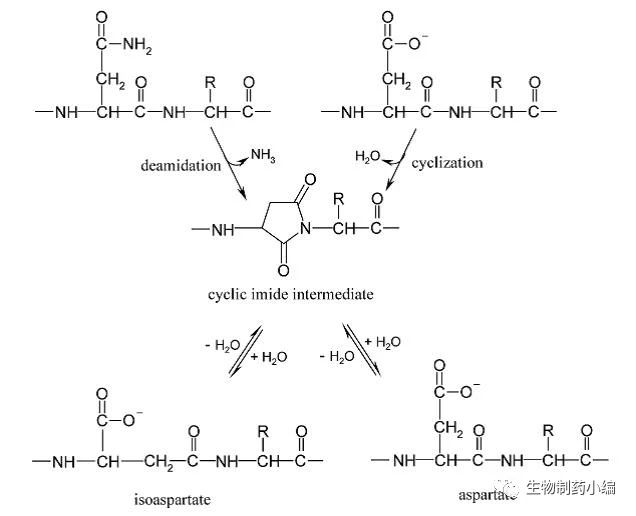
天冬酰胺残基的脱酰胺(Deamidation),天冬氨酸的异构化(Isomerization),以及环状琥珀亚酰胺(Succinimide)中间体的形成是蛋白药物在生产、储存及体内最为常见的化学降解反应,相关的化学反应机理见图2[6]。这些序列上的化学修饰可导致电荷异构体的生成和蛋白活性的丧失,甚至进一步造成蛋白的断裂(Cleavage)[7]。最常见、风险最高的脱酰胺发生位点的基序为Asn-Gly,另外在Asn-Ser/Thr/His等位置也常常发生,该反应在pH > 7环境中的发生速率很快。天冬氨酸异构化反应产生的基序则主要包括Asp-Gly/Ser/Asp等,该反应在pH < 6和高温时更易发生。在离子交换色谱的检测中,脱酰胺蛋白会形成酸性峰,琥珀亚酰胺形成碱性峰,异构化则视具体情况不同而不同。这些具体位点的鉴定可通过肽图-质谱来确认。
甲硫氨酸或色氨酸的氧化是蛋白分子另外一个主要的化学降解反应。造成蛋白氧化的活性氧可能来自光照、吐温和细胞培养基或设备中的金属离子。蛋白氧化除了可能会造成活性丧失、异质性增加,还可能进一步导致共价或非共价的聚集体形成,甚至产品变色。由于只有处于分子表面,暴露在溶剂环境中的甲硫氨酸/色氨酸才会有氧化风险,因此,仅基于序列一级结构的氧化位点分析可能与实验结果不能很好匹配。Adimab公司的一项研究对蛋白高级结构进行虚拟计算,建立了甲硫氨酸表面溶剂接触区域的计算模型,并进行了分子动力学模拟,对处于临床阶段的121个抗体均做了表面甲硫氨酸氧化位点的预测。同时也采用了过氧化氢处理这些抗体,强制使其氧化,并应用质谱对氧化位点进行了逐一鉴定。结果显示了虚拟预测和实际实验很好的相关性[8]。因此在成药性评价中采用这种虚拟预测方法可筛选大量候选分子,或对找到的位点进行突变以消除风险。
蛋白质的正确折叠是保持其生物学活性和稳定性的一个重要前提条件,且高级结构相对一级结构更为保守。高级结构的稳定性包括两个方面:一是蛋白分子本身维持正确折叠的构象稳定性(Conformational stability),二是正确折叠的蛋白分子与其自身在溶液中的相互作用形成的溶液整体稳定性,也称胶体稳定性(Colloidal stability)。由于高级结构的复杂性,虚拟计算手段目前还无法完全模拟分子在三维状态下各方面的各种作用力,因此蛋白高级结构稳定性的评价,目前还更多的依赖于实际的实验结果。
蛋白在高温或遇到化学变性剂时会发生去折叠,失去其高级结构,从而失去活性。通常使用差示扫描量热法(DSC)或差示扫描荧光法(DSF)对蛋白溶液进行梯度升温,来检测热变性温度(Tm),以反映其构象的热稳定性。其中前者通过测量蛋白变性过程所吸收的热量来反映,后者测量的是变性过程中疏水残基的暴露。另外,采用一系列浓度梯度的尿素或盐酸胍等化学变性剂促使蛋白变性,还可以通过其变性曲线计算,得到折叠蛋白和变性蛋白的能量差(ΔG)。
值得关注的是,多项研究发现,Tm和ΔG值与细胞培养表达量有高度的相关性。这两项数值越高,意味着蛋白构象越稳定,往往表达量也越高。在一项研究中发现,不同家族的抗体VL链的ΔG呈现出明显的区别,lambda链的ΔG为15-24 kJ/mol,而kappa链的为29-35 kJ/mol。与此对应,lambda片段的瞬时表达量为0.3-2 mg/l,而kappa片段可达5-17 mg/l[9]。另一项研究中,研究者将一个单抗的可变区进行了一些位点的组合突变,提高了其Tm和ΔG之后,发现表达量从低于1 mg/l骤然上升到了50 mg/l以上[10]。此外,近期的一项对处于临床阶段的137个单抗各种理化性质的聚类统计分析也表明,瞬时表达量与Tm高度相关[11]。这可能是由于蛋白在细胞内完成翻译之后,需正确折叠再分泌出胞外。构象稳定性较好的蛋白容易迅速折叠,并作为成核点促使更多的蛋白完成折叠。而易变性蛋白在细胞内变性堆积,则会形成细胞毒性,继而造成细胞活率下降。因此,Tm和ΔG值也成为成药性评价中最为常用的指标。
完整折叠的蛋白在溶液中会有自身相互作用,这种作用力主要由折叠后分子表面的疏水基团和电荷分布决定。当自身相互作用力表现为排斥力时,溶液就会表现出相对较好的胶体稳定性;而如果主要为吸引力时,就会容易产生聚集甚至沉淀,或表现为溶液粘度的增加。
如前文提到的,当分子表面呈现有聚成片的负电荷区域(negative patch),或疏水区域 (hydrophobic patch) 时,就会增加其自身间的相互吸引力。基因泰克的研究人员采用APBS和PDB2PQR电荷与溶剂化计算软件,对抗体表面电荷进行计算建模。将预测出的分子表面负电荷片区进行一些突变,去除负电荷聚集区后,蛋白溶液的粘度有了显著下降[12]。麻省理工和诺华的研究人员合作,研发出基于蛋白表面疏水片区作用预测蛋白聚集的SAP算法,继而又进一步整合入蛋白的净电荷分析和全长抗体结构分析,开发出成药性指数(DI)这一指标[13],目前已成为业界进行成药性评价常用的虚拟评价方法。
研究者们还开发了各种实验方法,来检测蛋白自身相互作用或表面疏水性质,以期用于成药性评价。基于静态光散射技术得到的第二维里系数(B22),和基于动态光散射技术得到的扩散相互作用指数(kD)均可用于指示分子间作用力大小。当这两个数值<0时,说明分子间为吸引力,数值越小说明吸引力越强;反之>0时则为排斥力[12]。AC-SINS法(Affinity-Capture Self-Interaction Nanoparticle Spectroscopy)和CSI-BLI法(Clone Self-Interaction by BioLayer Interferometry)也均可用来评估抗体自身相互作用,前者采用纳米金颗粒来实现,后者采用ForteBio的生物膜传感器实现。AC-SINS法还可在盐浓度梯度条件下进行(Salt-Gradient AC-SINS),则又可用来反映了抗体的疏水性质。另外两个用来反应表面疏水性质的方法,是疏水作用色谱(HIC)和直立单层吸附色谱(SMAC),前者可采用的市面上很多厂家提供的疏水作用色谱柱,后者主要采用Sepax公司的Zenix SEC-300色谱柱[11]。
值得一提的是,在以上所述的所有虚拟和实验的评价方法中,任何一项结果指示出的不理想蛋白性质,并不足以直接否定该分子成功商业化的可能。但一个分子所表现出的不及格指标越多,意味着后期失败的风险越大。因此,需要对所有评价方法得到的结果进行综合评估。
由于皮下注射在临床应用中的便捷性,很多治疗性单克隆抗体均希望开发成皮下注射制剂。但由于抗体药物剂量大,而皮下注射药液体积最多只能达1 ml,这就使得很多抗体制剂需要很高的蛋白浓度。一般来说,如果候选分子的预期制剂浓度高于50 mg/ml,则需要在成药性评价中考虑高浓度蛋白制剂的可行性。
溶解度和粘度是高浓度蛋白制剂可行性评价中主要考虑的两个因素。而影响这两个因素的内在分子机制主要还是由于分子表面的疏水或电荷作用。溶解度评估可简单采用超滤浓缩的方式将蛋白溶液持续浓缩,监测澄清溶液可达到的最大蛋白浓度;或研发早期蛋白量不够时采用PEG浓度梯度沉淀法对候选分子进行溶解度对比[14]。最近辉瑞的研究人员研发出一种虚拟计算的算法,可于早期用序列预测与浓度相关的粘度表现,也是基于分子表面电荷和疏水作用进行计算[15]。胶体稳定性评估方法,也可同样用于预估高浓度蛋白溶液的粘度表现。
在2014年之前,成药性评价更多的依赖于处方前研究工作,如极端pH、高温、冻融、光照、强制氧化等强制降解实验所产生的数据。这些工作需要非常多的样品量,且非常耗时。而在研发的早期阶段,又总是会面临可用样品量极少的情况。这就造成了项目的推进相对较慢,也需要更多的资源投入。
而近年来,随着人们对于蛋白分子结构越来越深入的研究,基于计算机虚拟计算的蛋白理化性质预测方法,结合所需样品量较少的快速评估实验方法,即可以极大的帮助研究者加速早期候选分子的筛选进程。在笔者看来,第一阶段通过这些高通量的评价方法,对大量的候选分子进行初步筛选;第二阶段再结合处方前研究工作,对筛选出的2 - 5个优选分子进行进一步评价和优化,则是蛋白药物发现中更有效率,成功率也更高的一种成药性评价策略。
[1] Reichert JM. Antibodies to watch in 2016. MAbs 2016, 8: 197-204.
[2] Lipinski CA. Lead-and drug-like compounds: the rule-of-five revolution. Drug Discov Today Technol 2004, 1:337-41.
[3] Jarasch A, Koll H, et al. Developability assessment during the selection of novel therapeutic antibodies. J Pharm Sci 2015, 104: 1885-98.
[4] Wei B, Berning K, et al. Glycation of antibodies: Modification, methods and potential effects on biological functions. MAbs 2017, 9: 586-594.
[5] Butko M, Pallat H, et al. Recombinant antibody color resulting from advanced glycation end product modifications. Anal Chem 2014, 86: 9816-23.
[6] Zhang W, Czupryn MJ. Analysis of isoaspartate in a recombinant monoclonal antibody and its charge isoforms. J Pharm Biomed Anal 2003, 30: 1479-90.
[7] Catak S, Monard G, et al. Computational study on non enzymatic peptide bond cleavage at asparagine and aspartic acid. J Phys Chem A 2008, 112: 8752-61.
[8] Yang R, Jain T, et al. Rapid assessment of oxidation via middle-down LCMS correlates with methionine side-chain solvent-accessible surface area for 121 clinical stage monoclonal antibodies. MAbs 2017, 9: 646-653.
[9] Rouet R, Lowe D, et al. Stability engineering of the human antibody repertoire. FEBS Lett 2014, 588: 269-77.
[10] Seeliger D, Schulz P, et al. Boosting antibody developability through rational sequence optimization. MAbs2015, 7: 505-15.
[11] Jain T, Sun T, et al. Biophysical properties of the clinical-stage antibody landscape. Proc Natl Acad Sci USA2017, 114: 944-949.
[12] Yadav S, Laue TM, et al. The influence of charge distribution on self-association and viscosity behavior of monoclonal antibody solutions. Mol Pharm 2012, 9: 791-802.
[13] Lauer TM, Agrawal NJ, et al. Developability index: a rapid in silico tool for the screening of antibody aggregation propensity. J Pharm Sci 2012, 101: 102-15.
[14] Li L, Kantor A, et al. Application of a PEG precipitation method for solubility screening: A tool for developing high protein concentration formulations. Protein Sci 2013, 22: 1118-23.
[15] Tomar DS, Li L, et al. In-silico prediction of concentration-dependent viscosity curves for monoclonal antibody solutions. MAbs 2017, 9: 476-489.


